

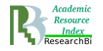



Introduction. GM1-gangliosidosis is a lysosomal disease of autosomal-recessive hereditary type, which is related to mutations in gene GLB1, encoding the protein of acid β-galactosidase. The general incidence rate of this disease in the world is 1:100,000–1:200,000 newborns [1, 391-396]. The mutations in gene GLB1 lead to the deficiency in acid β-galactosidase, and, as a result, the accumulation of GM1-gangliosidosis in the patients’ lysosomes, which triggers the development of a severe progressing neurodegenerative disorder. Taking into consideration the age of the disease manifestation and the severity of the clinical course, GM1-gangliosidosis is divided into three clinical forms: type I (infantile form), type II (juvenile form), type III (adult form). In patients with the infantile form, the disease is manifested during their first six months of life with severe pathology of the central nervous system, hepatosplenomegaly, and skeletal anomalies. The juvenile form is characterized by neurological changes and disorders in the locomotor system which have slow progression; the first symptoms of the disease are manifested at the age from 7 months to 3 years old. The adult form of the disease is manifested at the age from 3 to 30 years old and is characterized by the disorders in the coordination of movements, dystonia, developmental language disorders, short stature and moderate changes in the skeleton.
Gene GLB1 is mapped on chromosome 3 in locus 3р21-р14.2, is 62.5 kb long, consists of 16 exons and encodes 677 aminoacid residues [2, 403-404]. At present 144 mutations, associated with GM1-gangliosidosis, have been described for gene GLB1, 109 of which are missence/nonsense mutations, 14 – deletions, 10 – insertions, 11 – mutations, causing splicing disorders [3, 173-181]. The mutations in gene GLB1 are remarkable for high fluctuations in the frequency of specific mutations in different populations, there is no mutation, which could be considered major for many countries. However, there are some mutations, the frequency of which is high in some populations. In particular, in Brazil the mutation c.1622–1627insG among the studied patients with GM1-gangliosidosis was found in 50 % alleles, and the mutation p.R59H – in 19 % alleles. Contrary to the incidence rates for the world, the mutation c.1622–1627insG was described only for Brazilian patients, except for one patient from Uruguay [4, 113-116], the mutation p.R59H is common for Spanish patients (28 %) of Gipsy origin [5, 1060-1071].
Until today, there has been no analysis of the mutations in gene GLB1 for Ukrainian patients with GM1-gangliosidosis and their frequencies are unknown.
The aim of our work was to search for major mutations in GLB1 gene in Ukrainian patients with GM1-gangliosidosis.
Materials and Methods
The analysis of mutations in gene GLB1 was conducted in 26 patients from different regions of Ukraine, who visited the Medical Genetics Center (MGC) and the Center of Orphan Diseases (COD) of NCSH OKHMATDYT of the Ministry of Health of Ukraine to have a consultation regarding alleged presence of hereditary metabolic disorders. There was a preliminary diagnosis of GM1-gangliosidosis for the patients of the search group, determined by the results of the activity of acid β-galactosidase [6, 222-225]. The method of single strand conformational polymorphism of DNA fragments (SSCP) was used for primary screening of exons in GLB1 gene, which contain any changes [7, 3121-3128]. The final search for mutations was made by the Sanger’s method of direct automated sequencing of the required exon [8, 286] using ABI Prism 3100 device (Applied Biosystems) according to the manufacturer’s protocol. The selection of primers for each of 16 exons of gene GLB1 was conducted using Primer3 program, web-version 4.0.0 (http://bioinfo.ut.ee/primer3/). The analysis of sequencing results was made using programs Chromas and BLAST (http://www.ncbi.nlm.nih.gov/blast).
Results and discussion
The mutational analysis of gene GLB1 in 26 Ukrainian patients from different regions of Ukraine with the determined diagnosis of GM1-gangliosidosis revealed 15 pathogenic mutations (http://www.hgmd.cf.ac.uk), the majority of which were localized in exons 2, 6, 7, and 8 (the data of the studies have not been published yet). The most frequent mutation was found to be the missence-mutation p.His281Tyr (с.841С>T) in exon 8, which was found in at least one allele in 16 out of 26 patients (61.5 %), which was 20 out of 52 (38.5 %) mutant alleles of all the studied patients with GM1-gangliosidosis.This mutation was present in 4 patients in the homozygous state and 12 patients – in the heterozygous state. Such high frequency of this mutation allows considering it to be major for Ukraine.
The mutation p.His281Tyr (с.841С>T) in gene GLB1 was first described by Е. Paschke (2001) for one patient with GM1-gangliosidosis with severe neurological pathology [9, 159-166].
The analysis of the data, published in recent 10 years regarding the distribution of the mutation p.His281Tyr in other populations, was conducted by us (Table 1).
Table 1
Distribution of mutations p.His281Tyr (с.841С>T) in different populations.
| Сountry | The number of alleles with mutation p.His281Tyr of the general study of alleles | Frequency, % |
References |
| Ukraine | 20/52 | 38,5 | This work |
| Argentina | 0/38 | 0 | Santamaria R. et al., 2007 [10, 273-279] |
| Brazil | 0/130 | 0 | Fernanda Sperb et al., 2013 [4, 113-116] |
| Portugal | 2/28 | 7 | Coutinho MF et al., 2012 [11, 379-393] |
| Austria | 0/32 | 0 | Hofer D. et al., 2010 [12, 236-246] |
| Germany | 1/34 | 3 | Paschke E. et al., 2001 [9, 159-166] |
| Spain | 0/14 | 0 | Santamaria R. et al., 2007 [13, 2275-2282] |
| Italy | 2/50 | 4 | Caciotti et al., 2011 [14, 782-790] |
| UAE | 0/28 | 0 | Fatma A. et al., 2013 [15, 1-9] |
| Turkey | 0/10 | 0 | Başak Çeltikçi et al., 2012 [16, 571-574] |
| India | 0/100 | 0 | Bidchol, A.M., et al., 2015 [3, 173-181] |
| China | 0/10 | 0 | Yang et al., 2010 [17, 79-87], Hong-Lin Lei et al., 2012 [18, 359-362] |
The result of the analysis demonstrated that the mutation p.His281Tyr (с.841С>T) was described for single cases of patients from Germany, Italy, and Portugal, whereas no allele with this replacement was identified in other countries. This fact may testify to a considerably higher frequency of this mutation in Ukraine, compared to most countries.
The molecule of acid human β-galactosidase (EC 3.2.1.23, β-Gal) is a protein, consisting of three domains, which is in the monomer form at neutral pH (Fig. 1) [19, 1801-1812].
Fig.1 The domain organization of the human protein molecule β-Gal [19, 1801-1812]
In conditions of acid pH of the lysosome (рН 4–5) the molecule of human β-galactosidase acquires its active form by dimerization. The studies of the molecule structure of acid human β-galactosidase and the impact of mutations in gene GLB1 on it, conducted by Ohto et al., demonstrated that amino acid His281 is localized on the protein surface, and its replacement (in particular, in case of mutation p.His281Tyr) may result in the disorder of electrostatic properties of the surface of acid β-galactosidase protein, which may disturb the dimerization [19, 1801-1812]. Such structural and functional changes in the enzyme molecule lead to the decrease in the activity of this enzyme in lysosomes, and, as a result, in the occurrence of disorders, notable for GM1-gangliosidosis.
The level of activity of acid β-galactosidase in the patients with GM1-gangliosidosis, observed by us, was greatly decreased; it was from 0 to 4.2 % from the normal activity of the enzyme and was not different for the patients with the mutation p.His281Tyr, compared to other patients. The manifestation of the disease in the patients with this mutation occurred within the period from 3 to 6 months both for homo- and heterozygous state. Almost all the patients with this mutation had early infantile form of GM1-gangliosidosis with severe neurological symptoms. Only one patient with genotype p.His281Tyr/Arg201His had the adult form of GM1-gangliosidosis with milder clinical course, which may be conditioned by the softening effect of mutation Arg201His on the phenotype of the disease [20, 204-215].
The distribution frequency of mutation p.His281Tyr in the patients with GM1-gangliosidosis was analyzed by us in different regions of Ukraine. Fig. 2 demonstrates the map of Ukraine with the regions, where the patients with GM1-gangliosidosis, identified by us, live, and the number of alleles with the mutation p.His281tyr compared to the total number of mutant alleles, identified in this region.
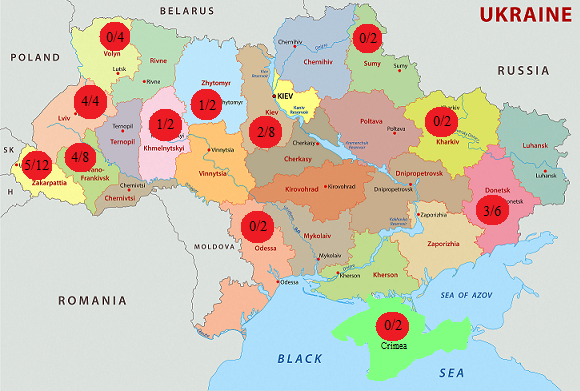
Fig. 3 Distribution of mutation p.His281Tyr (с.841С>T) in patients with GM1- gangliosidosis in regions of Ukraine
The analysis of the information about the territorial distribution of the identified families, suffering from the mutation p.His281Tyr (с.841С>T), demonstrated that most alleles with this mutation (14 out of 20, or 70 %) were identified in patients from western regions of the country. This high frequency of the mutation in the western regions of Ukraine may be conditioned by the founder effect [21, 592], related to a specific ethnic group of population due to geographic specificities of this territory with rather isolated settlements with small number of population, located at a great distance from each other.
Therefore, the mutational analysis of gene GLB1 in the Ukrainian patients with GM1-gangliosidosis demonstrated that, contrary to other populations, the missence-mutation p.His281Tyr (с.841С>T) is highly frequent for Ukrainian patients (38.5 %), thus it may be considered major for this population, first of all, for western regions of the country. These results may be used to elaborate the most rational algorithm of molecular diagnostics of GM1-gangliosidosis, using convenient and fast methods (allele-specific PCR, restrictional analysis) of identifying the mutation p.His281Tyr (с.841С>T), which would promote enhancing the quality and efficiency of GM1-gangliosidosis in Ukraine.